Kurs MPI - pracownia nr 3
Celem ćwiczenia jest nabranie biegłości w posługiwaniu się typami danych i operatorami definiowanymi przez użytkownika. Dokładny opis typów danych definiowanych przez użytkownika można znaleźć np. w rozdziale User-Defined Datatypes and Packing książki MPI: The Complete Reference
Studenci i doktoranci: kody źródłowe oraz wydruk otrzymanych wyników (najlepiej w postaci tar.gz) proszę wysłać mailem na adres adam@chem.univ.gda.pl; będzie to sprawozdanie z tej pracowni. Przysłanie samego tekstu programu nie zalicza pracowni. Czas: do 7 maja, 2008.
Wykorzystanie typów danych definiowanych przez użytkownika
Napisać program rozpraszający M cząstek
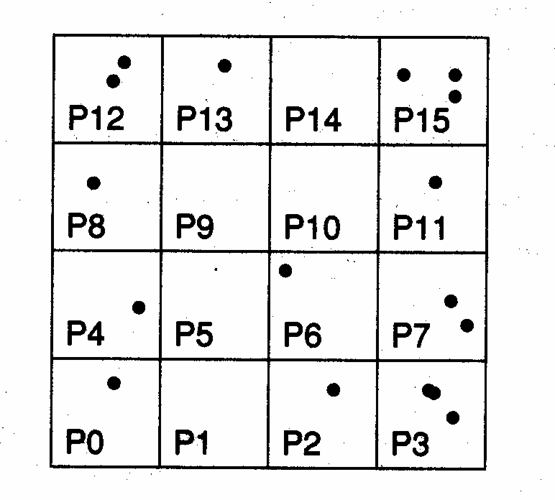
w kwadratowej komórce elementarnej pomiędzy N=(4,9,...,n2 )
procesorów w zależności od położenia w komórce zgodnie z rysunkiem (dla każdego
procesora P0...,P15 przypisujemy kwadratowy element wynikający z podziału
komórki elementarnej na N kwadratów jednakowych rozmiarów ):
Atom należy zdefiniować w postaci struktury o następujacych polach: nazwa atomu (łańcuch czteroznakowy), masa atomu (double), współrzędne kartezjanskie x, y (2 * double) i prędkości vx, vy (2 * double). W przypadku Fortranu 77 strukturze ma odpowiadac blok wspólny. Procesor P0 powinien wyznaczyć losowe położenia cząstek i rozesłać odpowiednie cząstki do kolejnych procesorów. Do rozwiązania zadania zdefiniować typy danych użytkownika pozwalające na wysyłanie wybranych elementów tablicy struktur do kolejnych procesorów.
Własne operatory redukcji dla MPI_REDUCE
Napisać program wyszukujący ze zbioru N atomów których dane są rozproszone pomiędzy N procesorów atomy o następujących charakterystykach:
(a) największej energii kinetycznej, gdzie Ekin i =1/2*mi*vi2
(b) największej odległości od początku układu współrzędnych
Dane te mają być zwracane do procesora o rzędzie 0. Do opisu każdego atomu przyjąć strukture danych o następujacych polach: nazwa atomu (łańcuch czteroznakowy), masa atomu (double), wspólrzędne kartezjanskie x, y, z (3 * double) i prędkości vx, vy, vz (3 * double). W przypadku Fortranu 77 strukturze ma odpowiadac blok wspólny.
Do rozwiazania zadania zdefiniowac przy pomocy procedury MPI_OP_CREATE dwa własne operatory redukcji MAX_EKN i MAX_DIST znajdujące odpowiednio atom o maksymalnej energii kinetycznej i atom o największej odleglości od początku układu współrzędnych.