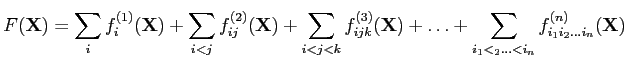The goal of my research is to understand how intra- and inter-molecular interactions make a given biomolecular system attain its unique structure and perform a given biological action. I use the coarse-graining approach for this purpose, in which interactions are treated at the site-site rather than detailed atomistic level. Because of a tremendous reduction of the number of degrees of freedom and, thereby, the CPU and memory requirements of computations, a practical application of this approach, which is pursued in my lab, is the development of methods with which to simulate the structure and dynamics of biomolecules and biomolecular systems at a large time and size scales, which are not accessible to all-atom simulations.
Together with my coworkers, I developed a systematic physics-based approach to coarse-graining, which is based on the decomposition of the potential of mean force (PMF) of a given system [1] into contributions due to its parts, thus enabling to produce a transferable force field. Based on this approach, we developed the UNRES model for the simulation of protein structure and dynamics [2,3], the NARES-2P model for nucleic acids [4] and the SUGRES-1P model for polysaccharides [5], which are now being merged into the Unified Coarse-Grained Model [5]. We are currently working on the extension of the model to incorporate multi-scale approach, where some parts of a system are treated at the all-atom level.
UNRES, NARES-2P, and SUGRES-1P are minimalistic models of the respective biological macromolecules, with one or two sites per residue. Such a representation has resulted in an over 1000-fold speed-up of computations with respect to the all-atom approach [6]. Yet, owing to the physics-based principles of force-field construction, the model is able to predict protein structure [4], reproduce protein-folding kinetics [7] and free-energy landscapes [8], simulate DNA and RNA structure, dynamics, and thermodynamics [9], and simulate biologically-important processes [10,11]. It can also handle D-amino-acid residues [12] and dynamic formation of disulfide bridges [13]. The UNRES component of the model is available from the www.unres.pl web page and has recently also been implemented on the Polish Computer Grid Network (PL-GRID) as the UNRES-NG utility. The current version of NARES-2P can fold small RNA molecules and hybridize small DNA molecules, as well as reproduce well the thermodynamical parameters of folding of small DNA molecules and reproduce ``bubble'' formation (or pre-melting transition) in AT-rich sequences [9].
In our approach [1], the prototype of the effective energy
function of a system is the potential of mean force (PMF), also termed restricted energy function
(RFE) of the system of interest,
![]() , where
, where
![]() is a shorthand for the
coarse-grained degrees of freedom. All degrees of freedom that are lost when passing from the all-atom
to the coarse-grained model (
is a shorthand for the
coarse-grained degrees of freedom. All degrees of freedom that are lost when passing from the all-atom
to the coarse-grained model (
![]() ) are averaged over, as expressed by Eq. (1).
) are averaged over, as expressed by Eq. (1).
where
![]() ,
,
![]() is the original (all-atom) energy function,
is the original (all-atom) energy function,
![]() is the universal gas constant,
is the universal gas constant, ![]() is the absolute temperature,
is the absolute temperature,
![]() is the region of the
is the region of the
![]() subspace of variables over which the integration is carried out, and
subspace of variables over which the integration is carried out, and ![]() is the
volume of this region. The PMF is decomposed into factors of increasing order [Eq. (2)];
factors of order 1 (
is the
volume of this region. The PMF is decomposed into factors of increasing order [Eq. (2)];
factors of order 1 (![]() pertain to interactions withing single coarse-grained sites and pairs of
sites, while factors of higher order correspond to clusters of sites and give rise to multibody terms.
pertain to interactions withing single coarse-grained sites and pairs of
sites, while factors of higher order correspond to clusters of sites and give rise to multibody terms.
A tremendous advantage of this approach compared to statistical and other physics-based potentials [14,15] is that the total potential of mean force of the system is split, in a rigorous way, into a sum of contributions from its smaller parts, which enables us to construct a transferable force field, and that analytical expressions for the terms in the simplified energy function, including the multibody terms essential to maintain regular structure [16,1], can be obtained [1] by the expansions of the factors into Kubo's cluster cumulants [17].
Use of the approach outlined above for a specific system/class of systems involves the following steps [2,5]: (i) determination of coarse-grained sites and determination of the maximum order of the factor expansion; (ii) derivation of approximate analytical expressions for the factors by Kubo's cumulant expansion [17] or other means; (iii) computing the surfaces of the factors based on the PMF surfaces of model systems calculated by direct integration or all-atom MD simulations, and (iv) calibration of the force field to reproduce the structure and thermodynamical properties of training molecules. This approach was followed when developing the Unified Coarse Grained Model.
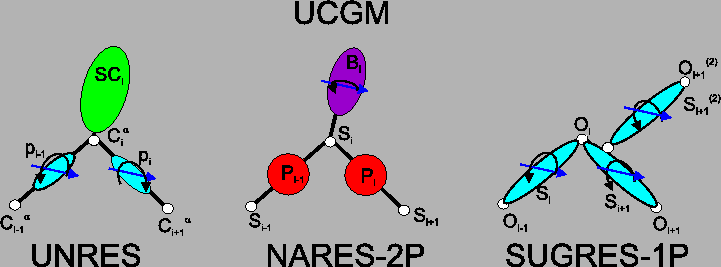
The components of the Unified Coarse Grained Model of biological macromolecules are schematically depicted in Figure 1 [2,4,5]. Their common feature is the presence of an interaction site capable of hydrogen bonding, namely the peptide group (blue spheroids in the left panel of the Figure) for UNRES, united sugar-base groups (purple spheroids in the middle panel of the Figure) for NARES-2P, and sugar rings, which are the only interaction sites in the SUGRES-1P model (blue spheroids in the right panel of the Figure).
 |
To use the model in molecular simulations, the Langevin molecular dynamics approach [6] and its multiplexed replica-exchange (MREMD) and multicanonical molecular dynamics (MUCAMD) were implemented [18,19].
The current focus in the development of NARES-2P and SUGRES-1P is directed at the derivation of local potentials from the PMF surfaces of model systems obtained by all-atom MD simulations. Analytical expressions are fitted to these surfaces as in previous work [20,12]. UNRES is now being enhanced with a mean-field model of a lipid membrane.
My coworkers and I have recently developed a novel approach to force-field calibration, which
is based on maximum-likelihood fitting of the conformational ensembles
generated by simulations to the ensembles determined experimentally at
various temperatures by means of the maximum-likelihood principle [21].
The method was tested with tryptophan cage as the training protein for which
NMR structures were determined at three temperatures, including the
folding-transition temperature [22] and found to be very
stable and to produce a force field transferable with which to predict the
structures of ![]() -helical proteins [21].
Calibration of UNRES with
-helical proteins [21].
Calibration of UNRES with ![]() -,
-,
![]() -, and
-, and ![]() -proteins
is in progress.
-proteins
is in progress.
The new method was also applied to calibrate NARES-2P [23].
The target function was supplemented with the ![]() terms corresponding
to the difference between the calculated and the experimental heat-capacity
curves. The resulting force field reproduced the thermodynamics of DNA
hybridization better than the original NARES-2P force field.
terms corresponding
to the difference between the calculated and the experimental heat-capacity
curves. The resulting force field reproduced the thermodynamics of DNA
hybridization better than the original NARES-2P force field.
UNRES is being tested in every Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP). To enhance its predictive power, my coworkers and I have recently designed and tested an approach in which restraints from templates [24] or other knowledge-based sources such as, e.g., contact prediction [25] are used as restraints in UNRES. This approach is particularly successful when individual domains are well predicted by template-based modeling while their packing is not because of poor similarity scores in the linker region [24].
With Dr. Antti Niemi, we are investigating protein folding pathways in terms of the dark-soliton solutions of Discrete Nonlinear Schrödinger equation. Dark solitons are identified in the folding trajectories obtained by coarse-grained simulations with UNRES and soliton evolution is studied. With this approach, we are studying the folding of staphylococcal protein A [26].
My coworkers and I are currently studying the free-energy landscapes [8] and kinetics of the folding the Fbp28 WW domain and its mutants [7]. The rate constants determined by fitting kinetic equations to the simulated profiles of the folded and intermediate structures turned out to correlated well with the experimental constants. The treatment is now being extended to include hydrodynamic interactions in the Langevin dynamics scheme of UNRES simulations.
My coworkers and I are working on the investigation of the mechanism of conformational transition of Hsp70 chaperones; we simulated the structure of bacterial Hsp70 [10] before the experimental structure was determined [27]. We are also studying the conformational changes of Hsp70 at the atomic level [28]. Our current research is focused on the influence of chaperone conformation on peptide-substrate binding. We are also working iron-sulfur cluster biogenesis [11]. We proposed the most probable binding modes of the iron-sulfur 1 (Isu1) protein to the Jac1 protein and are now extending the approach to the ternary Ssq1/Isu1/Jac1 complex, where Ssq1 is the Hsp70 chaperone from yeast.
The immediate objective of my research is introduction multiscale treatment of the systems under investigation to the Unified Coarse Grained Model, in which the most of the system is treated at the coarse-grained level and parts of interest (e.g., binding sites and ligands) at the detailed atomic level. Because the prototype of the energy function of the model is the potential of mean force, the potentials of mean force due to the parts treated at atomic level will be calculated on the fly during simulation and used to compute their interactions with the coarse-grained part. This approach will enable to consider metal ions, small ligands and the docking of which cannot be handled at the coarse-grained level because of too low resolution of this treatment. I also plan to combine this approach with the QM/MM treatment to enable the consideration of allostery in studying enzymatic reactions, proton and electron transfer, etc.
In the development of UNRES and NARES-2P, studying protein-folding kinetics and
free-energy surfaces, and applying the coarse-grained model to solve
biological problems, I have an established long-term
collaboration with Dr. Harold A. Scheraga, Cornell University, U.S.A.,
Dr. Stan Oldziej, University of Gda![]() sk and Medical University of Gda
sk and Medical University of Gda![]() sk,
Poland, and Dr. Maciej Kozak, Adam Mickiewicz University, Pozna
sk,
Poland, and Dr. Maciej Kozak, Adam Mickiewicz University, Pozna![]() , Poland.
In using knowledge-based information in UNRES, I am collaborating with
Dr. Jooyoung Lee, Korea Institute for Advanced Study, Seoul, Republic
of Korea, and Dr. Silvia Crivelli, National Instituted of Health, U.S.A.,
and Dr. Michela Taufer, Delaware University, U.S.A. I am also collaborating
with Dr. Antti Niemi, Uppsala University, Sweden, University of Tours,
France and Beijing Institute of Technology (BIT), P.R. China, and with Dr. Jianfeng
He, BIT, and Dr. Yi Xiao, Huazhong University of Science and Technology,
Wuhan, P.R. China, in using UNRES and the Landau Hamiltonian to study protein folding.
, Poland.
In using knowledge-based information in UNRES, I am collaborating with
Dr. Jooyoung Lee, Korea Institute for Advanced Study, Seoul, Republic
of Korea, and Dr. Silvia Crivelli, National Instituted of Health, U.S.A.,
and Dr. Michela Taufer, Delaware University, U.S.A. I am also collaborating
with Dr. Antti Niemi, Uppsala University, Sweden, University of Tours,
France and Beijing Institute of Technology (BIT), P.R. China, and with Dr. Jianfeng
He, BIT, and Dr. Yi Xiao, Huazhong University of Science and Technology,
Wuhan, P.R. China, in using UNRES and the Landau Hamiltonian to study protein folding.
My current research is supported by two grants from the Polish National
Science Center and Foundation for Polish Science. I have access to the
supercomputers of the Academic Computer Center in Gda![]() sk TASK
(www.task.pl), Interdisciplinary Center of Mathematical and Computer Modeling,
University of Warsaw (www.icm.edu.pl) and, through collaboration with H.A. Scheraga
and S. Crivelli, to XSEDE and NERSC supercomputing facilities in the U.S.A.
sk TASK
(www.task.pl), Interdisciplinary Center of Mathematical and Computer Modeling,
University of Warsaw (www.icm.edu.pl) and, through collaboration with H.A. Scheraga
and S. Crivelli, to XSEDE and NERSC supercomputing facilities in the U.S.A.
![$\displaystyle F({\bf X})=-RT\ln{\biggl\{\frac{1}{V_{\bf Y}}\int_{\Omega_{\bf Y}}\exp[-E({\bf X};{\bf Y})/RT]} {\rm dV}_{\bf Y}\biggr\}$](img4.png)